|
Russell H. Swerdlow, MD Gene and Marge Sweeney Professor Professor of Neurology, Cell Biology and Physiology, Biochemistry & Molecular Biology Director, University of Kansas Alzheimer's Disease Research Center Director, KUMC Neurodegenerative Disorders Program University of Kansas School of Medicine Dr. Swerdlow continues his research into Alzheimer’s disease (AD) mitochondrial dysfunction. Studies focus on genes that underly mitochondrial function including mitochondrial DNA (mtDNA) and the APOE gene; mitochondrial connections to the classic AD pathologies including amyloid plaques and tau tangles; and how to manipulate mitochondria to treat AD patients. Efforts advance Dr. Swerdlow’s “Mitochondrial Cascade Hypothesis,” which uniquely proposes AD arises as a primary consequence of mitochondrial stress. According to this novel paradigm, mitochondria initiate AD, drive its progression and pathologies including plaques and tangles, and provide a roadmap for developing effective therapies. Figure 1 illustrates the program’s overall organization: 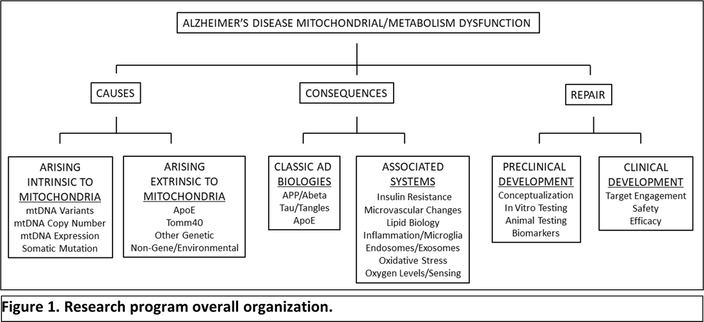 Stop Alzheimer’s Now support was especially critical in supporting research performed by one of Dr. Swerdlow’s graduate students, Scott Koppel MD PhD. Dr. Koppel is now pursuing his neurology residency training at Cedars Sinai Medical Center in Los Angeles. Stop Alzheimer’s Now is acknowledged as a major benefactor of the following publication, which found that a ketogenic diet affects the brain in ways that may be expected to counter brain changes that are seen in Alzheimer’s disease patients:
Of note, in June 2022 Dr. Swerdlow received an international award, the Oskar Fischer Prize. The awarding committee recognized the Mitochondrial Cascade Hypothesis as one of the world’s most promising attempts to conceptualize AD. In the past year Dr. Swerdlow delivered invited Mitochondrial Cascade Hypothesis national and international presentations to these forums:
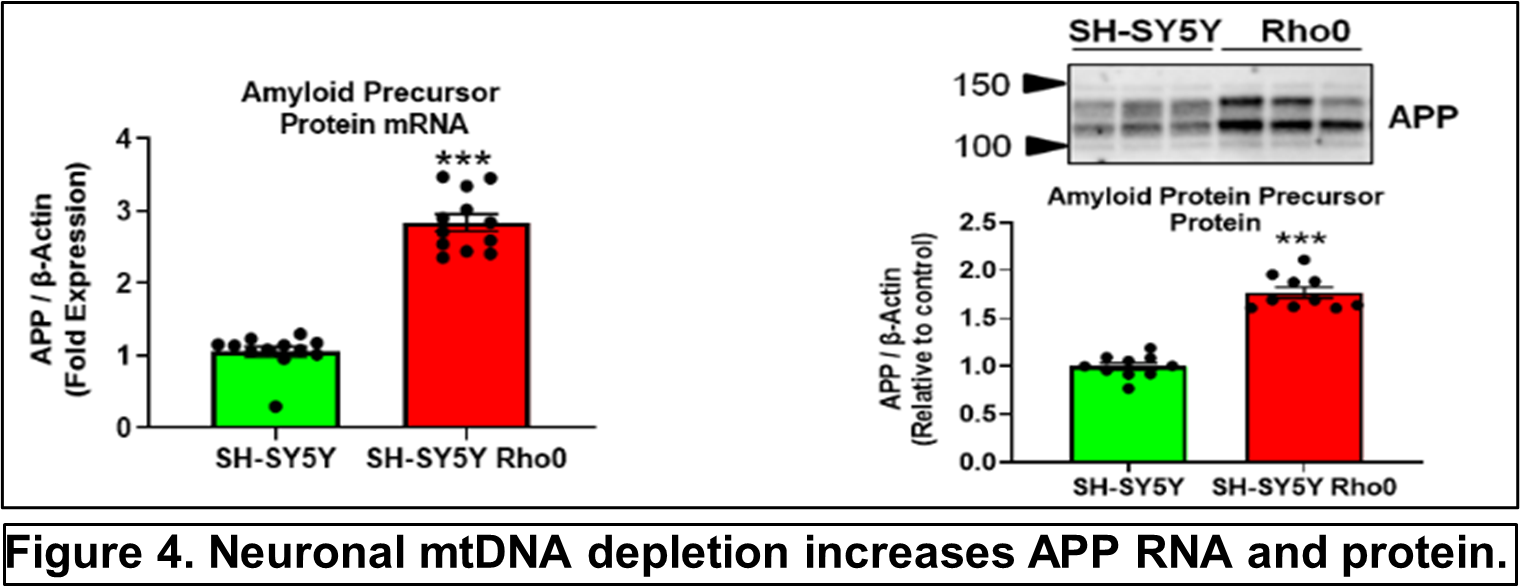
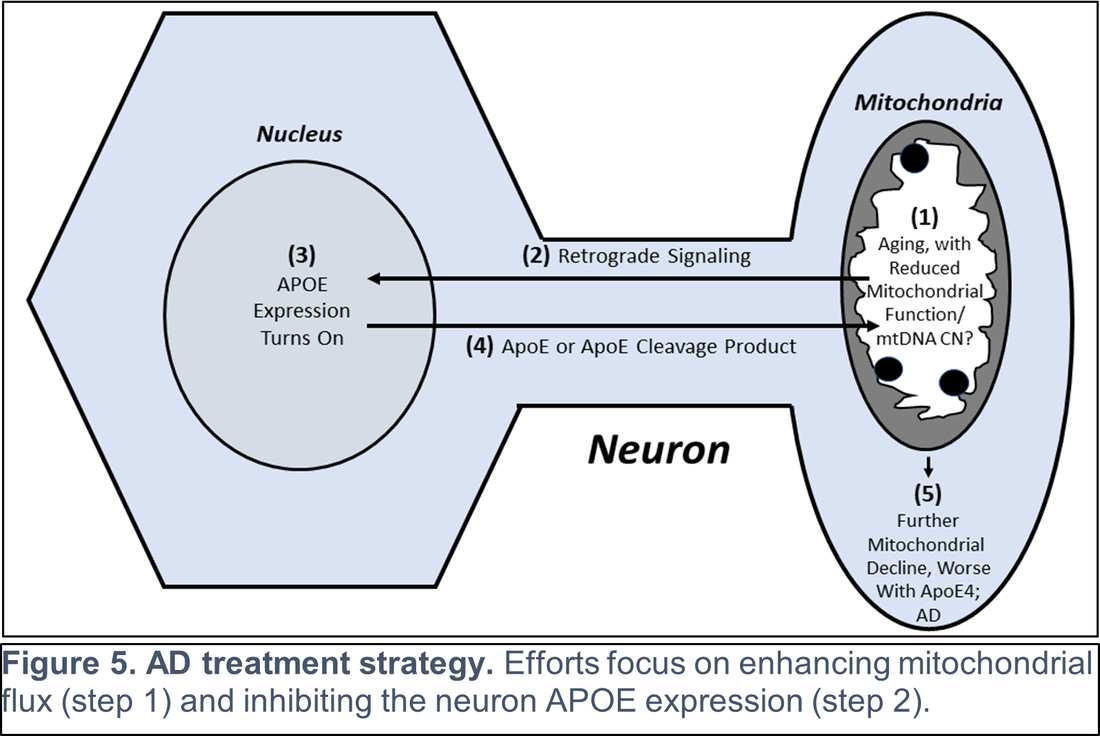
Other recent publications can be found here. Specific recent major accomplishments, and immediate directions, are discussed below. Genetic Studies We reported mitochondrial DNA (mtDNA) copy number was lower in brains of persons who died with AD than it was in the brains of persons who did not. We further demonstrated inverse associations between mtDNA copy number and AD histology changes (plaques and tangles) and the magnitude of cognitive impairment within an individual. Essentially, brains containing less mtDNA contained more plaque and tangle pathology, and persons with less mtDNA had more cognitive impairment. Some investigators will assume reduced mtDNA copy number is a consequence and not a cause of AD. We suspect it is more likely a cause and data we generated support this view. We found a specific heritable mtDNA sequence, haplogroup J, associates with higher mtDNA copy number and increased plaque/tangle pathology. Haplogroup J carriers also show evidence of reduced late-life memory capacity, which manifests prior to AD symptoms. In a related study, we observed aging mice normally upregulate their brain mtDNA copy number. Given this perspective, we propose that to maintain normal function neurons containing haplogroup J mtDNA must maintain a higher baseline mtDNA copy number, and as those neurons age their capacity to mount an age-determined compensatory increase in their mtDNA is consequently diminished. The APOE gene that encodes the apolipoprotein E protein associates with AD for unclear reasons. We find that in settings of mitochondrial dysfunction, neurons turn on APOE gene expression. This is important because neurons normally do not express APOE. We subsequently examined AD autopsy brains to look for evidence of neuron APOE expression and found APOE-laden neurons (Figure 2). This is remarkable because APOE is a secreted protein, so in addition to aberrantly expressing APOE the neurons appear unable to properly secrete it. 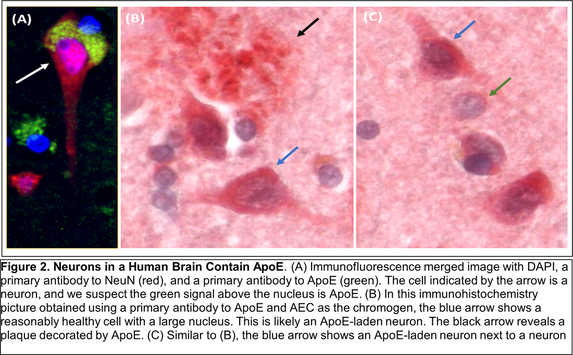 Linking Mitochondria to AD Pathologies Our recently published data show when neuronal cells express the amyloid precursor protein (APP), only a small amount localizes to mitochondria. Most APP distributes to the plasma membrane. Neurons process APP to beta amyloid (Aβ), which they secrete. We found that in settings of mitochondrial dysfunction, APP redirects to mitochondria, less reaches the plasma membrane, and less Aβ secretion occurs. We believe this can at least partly explain why cerebrospinal (CSF) Aβ levels are reduced in AD patients. We in fact believe CSF levels can serve as a biomarker of brain mitochondrial function. To address this hypothesis, we recently began obtaining CSF from comatose patients hospitalized in the KU Hospital intensive care unit with traumatic brain injuries or subarachnoid hemorrhage. Brain mitochondria in comatose patients are relatively inactive, but activity returns as the coma resolves. We are testing for associations between level of consciousness and Aβ in these samples. We are also measuring the tangle-forming protein tau, as well as mitochondrial components including mtDNA. Through this we hope to develop CSF assays that will inform the status of a living person’s brain mitochondria without having to perform an actual brain biopsy. We are further working to link mitochondrial and APOE biology. We are interested in how neuronal apolipoprotein E impacts overall neuron integrity, and directly or indirectly affects mitochondrial function. We therefore created neuronal cell lines that uniquely express the different APOE2, 3, and 4 isoforms. We predicted APOE4, the isoform that associates with increased AD risk, would disproportionally impair mitochondria but surprisingly discovered constitutive expression of all APOE isoforms induces equivalent states of mitochondrial stress. The apolipoprotein E protein plays a recognized role in lipid biology, which is broadly altered in AD patients. Accordingly, we initiated a “lipidomics” study to determine how mitochondrial dysfunction alters neuronal cell lipidomes.
Mitochondrial and Metabolism-Targeted AD Therapeutics We recently enrolled our 30th participant into a randomized trial that is evaluating how a ketogenic diet affects cognitive abilities in AD patients. Our goal is to is matriculate 80 participants. The rationale is that inducing ketone body production through a ketogenic diet will partly compensate for the state of reduced glucose utilization we know exists in AD. We believe this state reflects a consequence of mitochondrial dysfunction, and that adding the ketone fuel will enhance the brain’s ability to generate energy and signals that will promote mitochondrial maintenance. As part of this study, we perform extensive assays using blood cells from our trial participants. This includes applying a powerful omics technique, RNASeq, to determine how a ketogenic diet effects overall gene transcription in the trial participants. This past year we also completed a study assessing the impact of a diabetes drug, dapagliflozin, on AD patients. Dapaglifozin, an SGLT2 inhibitor, blocks the local re-uptake of renally excreted glucose. This leads to a mild reduction in blood glucose levels and a mild state of ketosis. We are currently analyzing the dapagliflozin trial data. In 2020 we reported that administering adequate doses of the metabolism intermediate oxaloacetate (OAA), a dicarboxylic acid, can promote brain bioenergetic fluxes in human AD patients. Because OAA is unstable in solution, we created OAA prodrugs in which OAA’s carboxyl ends are esterified to a ketone body, β-hydroxybutyrate, or a lactate precursor, propylene glycol. The former molecule was designed to deliver OAA as part of a ketogenic diet mimetic intervention, and the latter to deliver OAA as part of an exercise mimetic intervention. We are currently determining oral absorption, tissue distribution, and pharmacokinetics in the preclinical setting. These studies, which required us to generate isotope-labeled versions of the compounds, are intended to move us towards investigational new drug application (IND) filings with the FDA.
Support from Stop Alzheimers Now dramatically accelerated the pace and elevated the quality of Dr. Swerdlow’s research program. Since his initial support, the lab has enhanced its international recognition and reputation. Stop Alzheimers Now support was instrumental to our successes and is sincerely appreciated.
0 Comments
Leave a Reply. |